Research
We use tools derived from field biology, molecular immunology, and epidemiological modeling to study how zoonotic pathogens persist, transmit, and evolve in wild reservoir and spillover human hosts. Much of our work is focused on zoonotic infections derived from bat reservoirs, and we conduct the majority of our field studies in Madagascar. Major areas of research in the lab are summarized below.
Why are bat viral zoonoses so virulent?
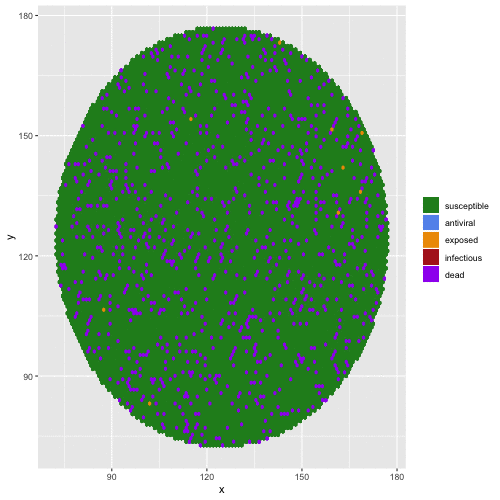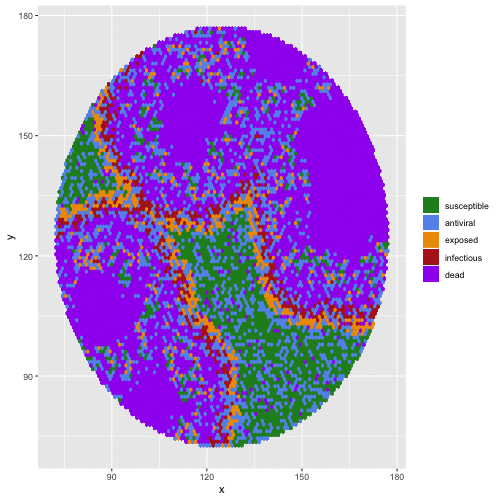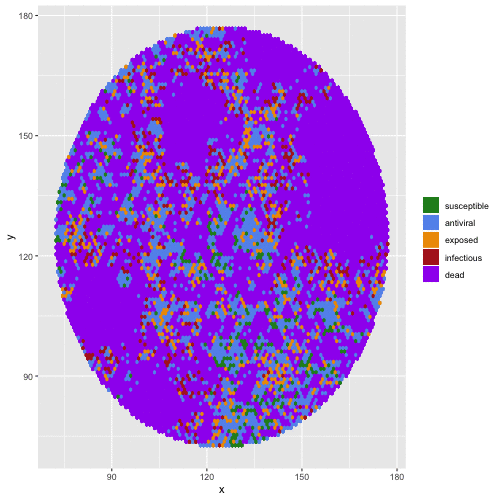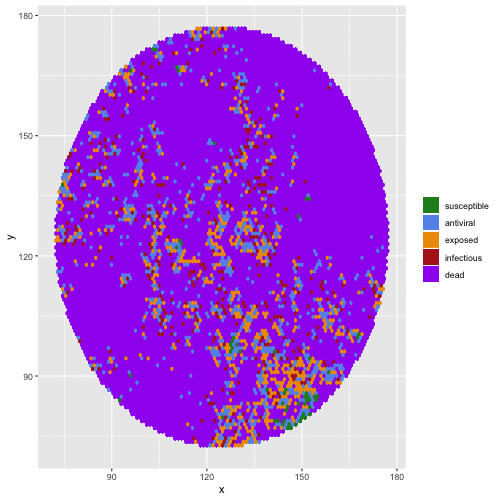
Bats are reservoir hosts for the world’s most virulent emerging viral zoonoses, including Hendra and Nipah henipaviruses, Ebola and Marburg filoviruses, and SARS, MERS, and SARS-CoV-2 coronaviruses. Amazingly, they are able to host these infections without themselves demonstrating substantial clinical pathology. Accumulating molecular evidence links the evolution of flight to bat viral tolerance: unique anti-inflammatory pathways in bat immune systems enable them to avoid oxidative damage accrued during the metabolically expensive process of flight, with cascading consequences for bat longevity and resilience to intracellular infections. Bats’ abilities to host viruses without disease may provide selective pressure for the evolution of pathogens which are not virulent to their bat hosts but cause extreme disease upon spillover to non-bat (i.e. human) hosts. We use a combination of experiments carried out in bat cell tissue culture and theoretical models to investigate the co-evolution of bat immune systems and their pathogens.
- Brook CE, Rozins C, Guth S, Boots M. 2023. Reservoir host immunology and life history shape virulence evolution in zoonotic viruses. PLoS Biology. 21 (9): e3002268. doi: 10.1371/journal.pbio.3002268.
- Guth S, Mollentze N, Renault K, Streicker DG, Visher E, Boots M, and Brook CE. 2022. Bats host the most virulent—but not the most dangerous—zoonotic viruses.PNAS. 119 (14): e2113628119. doi: 10.1073/pnas.2113628119.
- Brook CE, Boots M, Chandran KC, Dobson AP, Drosten C, Graham AL, Grenfell BT, Müller MA, Ng M, Wang LF, and van Leeuwen A. 2020. Accelerated viral dynamics in bat cell lines, with implications for zoonotic emergence. eLife. 9: e48401. doi: 10.7554/eLife.48401.
- Brook CE and Dobson AP. 2015. Bats as ‘special’ reservoirs for emerging zoonotic pathogens. Trends in Microbiology. doi: 10.1016/j.tim.2014.12.004.
How are viruses maintained in wild bat hosts?

The Brook Lab investigates how highly virulent viruses with acute infectious periods are maintained at the population-level in wild bat reservoir hosts. Classic epidemiological modeling explores the dynamics of perfectly immunizing childhood infections, under which hosts are born susceptible (S), become infectious (I), and recover (R) via immune responses retained for life. In the simplest example, infections are maintained in a population by constant re-supply of susceptible births. Many bat species reproduce in annual or biannual birth pulses, meaning that susceptible re-supply is restricted within a year. Such a system suggests that bat viruses may be governed by more nuanced dynamics than standard SIR–be they latent periods for persistent infections, periodic waning immunity, or metapopulation structure. To elucidate these dynamics, we fit candidate mechanistic models to age-structured prevalence and seroprevalence data, which we collect ourselves through the field studies of the inimitable Association Ekipa Fanihy.
- Madera S, Kistler A, Ranaivoson HC, Ahyong V, Andrianiaina A, Andry S, Raharinosy V, Randriambolamanantsoa TH, Ravelomanantsoa NAF, Tato CM, DeRisi JL, Aguilar HC, Lacoste V, Dussart P, Heraud JM, and Brook CE. 2022. Discovery and genomic characterization of a novel henipavirus, Angavokely virus, from fruit bats in Madagascar. Journal of Virology. 96 (18): e00921-22. doi: 10.1128/jvi.00921-22.
- Kettenburg G, Kistler A, Ranaivoson HC, Ahyong V, Andrianiaina A, Andry S, DeRisi JL, Gentles A, Raharinosy V, Randriambolamanantsoa TH, Ravelomanantsoa NAF, Tato CM, Dussart P, Heraud JM, and Brook CE. 2022. Full genome Nobecovirus sequences from Malagasy fruit bats define a unique evolutionary history for this coronavirus clade. Frontiers in Public Health. doi: 10.3389/fpubh.2022.786060.
- Gentles A, Guth S, Rozins C, and Brook CE. 2020. A review of mechanistic models of viral dynamics in bat reservoirs for zoonotic disease. Pathogens and Global Health. doi: 10.1080/20477724.2020.1833161.
- Brook CE, Ranaivoson HC, Broder CC, Cunningham AA, Héraud JM, Peel AJ, Gibson L, Wood JLN, Metcalf CJE, and Dobson AP. 2019. Disentangling serology to elucidate henipa- and filovirus transmission in Madagascar fruit bats. Journal of Animal Ecology. doi: 10.1111/1365-2656.12985.
What are the dynamics of zoonotic pathogens across the animal-human interface?

The Brook lab also undertakes several research projects that aim to understand the dynamics of pathogens during and after spillover from wildlife reservoirs to human populations. During the COVID-19 pandemic, we were awarded a Bill & Melinda Gates Foundation Grand Challenges Explorations grant which supported the establishment of the first-ever Illumina sequencing platform in Madagascar, through which we contributed >1000 Malagasy SARS-CoV-2 genomes to the global repository, GISAID. Currently, we have several Next Generation Sequencing and serosurveillance projects with applied human health focus underway in Madagascar, including one that is explicitly aimed at identifying the etiology of human fevers, with the idea of uncovering previously unrecognized zoonoses. More recently, we have also begun collaborating with researchers at the NIH ICER program in Cambodia–both in transmission inference of surveillance data for human pathogens (chiefly dengue), as well as in some exploratory zoonotic exposure work.
- Brook CE, Rozins C, Bohl JA, Ahyong V, Chea S, Fahsbender E, Huy R, Lay S, Leang R, Li Y, Lon C, Man S, Oum M, Northrup GR, Oliveira F, Pacheco AR, Parker DM, Young KI, Boots M, Tato CM, DeRisi JL, Yek C, and Manning JE. Climate, demography, immunology, and virology combine to drive two decades of dengue virus dynamics in Cambodia. In Review. Link to MedRxiv Preprint
- Tegally H, San J, Cotten M, Tegomoh B, Martin D...Brook CE...Ranaivoson HC...and Wilkinson E (397 authors). The evolving SARS-CoV-2 epidemic in Africa: Insights from rapidly expanding genomic surveillance. 2022. Science. 378 (6615): eabq5358. doi: 10.1126/science.abq5358
- Wilkinson E, Giovanetti M, Tegally T, San JE, Lessels R, Cuadros D, Martin DP, Zekri A-RN, Sangare AK, Ouedraogo A-S, Sesay AK, Hammami A, Amuri AA, Sayed A, Rebai A, Elargoubi A, Trotter AJ, Keita AK, Sall AA, Kone A, Souissi A, Gutierrez AV, Page AJ, Iranzadeh A, Lambisia A, Sylverken A, Ibrahimi A, Dhaala B, Kouriba B, Kleinhans B, Brook CE...Ranaivoson HC... and de Oliveira T (291 authors). A year of genomic surveillance reveals how the SARS-CoV-2 pandemic unfolded in Africa. 2021. Science. 374 (6566): 423-431. doi: 10.1126/science.abj4336
- Andriamandimby SF*, Brook CE*, Razanajatovo N, Rakotondramanga J-M, Rasambainarivo F, Raharimanga V, Razanajatovo IM, Mangahasimbola R, Razafindratsimandresy R, Randrianarisoa S, Bernardson B, Rabarison JH, Randrianarisoa M, Nasolo FS, Rabetombosoa RM, Randremanana R±, Heraud J-M±, Dussart P±. 2022. Cross-sectional cycle threshold values reflect epidemic dynamics of COVID-19 in Madagascar. Epidemics. 38: 1000533. doi: 10.1016/j.epidem.2021.100533.
What are the population viability trajectories of threatened animal species in Madagascar?

The compartmental modeling tools that the Brook lab employs above to address questions about the dynamics of pathogens are equally applicable to studies of viability and persistence for the populations that host them (e.g. bats). In the past, using age data derived from fruit bat dentition, we have undertaken population modeling studies for wild, Madagascar fruit bats. These studies suggest that the Madagascar Flying Fox, Pteropus rufus, is particularly threatened by habitat destruction and human hunting for food consumption in Madagascar. With funding from the Walder Foundation, we are currently working with a local conservation NGO, Madigasikara Voakajy, to (a) quantify P. rufus population connectivity through the use of GPS transmitters, (b) undertake new studies aimed at quantifying the positive benefits that this bat offers to healthy ecosystems (including pollination of several endangered Malagasy baobabs), and (c) develop conservation-oriented intervention plans to safeguard this species’ future. We are also involved in several other conservation collaborations, through which we have undertaken population modeling for several non-Chiropteran threatened taxa, including endemic Malagasy lemurs and tenrecs.
- Andrianiaina A, Andry S, Gentles A, Guth S, Héraud JM, Ranaivoson HC, Ravelomanantsoa NAF, Treuer T, and Brook CE. 2022. Reproduction, seasonal morphology, and juvenile growth in three Malagasy fruit bats. Journal of Mammalogy . doi: 10.1093/jmammal/gyac072.
- Annapragada A, Brook CE, Luskin MS, Rahariniaina RP, Helin M, Razafinarivo O, Ralaiarison AR, Randriamady HJ, Olson LE, Goodman SM, Golden CD. Evaluation of tenrec population viability and potential sustainable management under hunting pressure in northeastern Madagascar. 2021. Animal Conservation. doi: 10.1111/acv.12714.
- Brook CE, Ranaivoson HC, Andriafidison D, Ralisata M, Razafimanahaka J, Héraud JM, Dobson AP, and Metcalf CJE. 2019. Population trends for two Malagasy fruit bats. Biological Conservation. 234: 165-171. doi: 10.1016/j.biocon.2019.03.032.
- Brook CE, Herrera JP, Borgerson C, Fuller E, Andriamahazoarivosoa P, Rasolofoniaina BJR, Randrianasolo JLRR, Rakotondrafarasata ZRE, Randriamady HJ, Dobson AP, and Golden CD. 2018. Population viability and harvest sustainability for Madagascar lemurs. Conservation Biology. doi: 10.1111/cobi.13151.